
Abstrak
Biomassa memiliki kemampuan unik untuk menangkap CO2 atmosfer , meskipun untuk sementara waktu karena dekomposisi akhirnya melepaskan jumlah CO2 yang setara . Mengubah biomassa menjadi hydrochar menawarkan cara yang menjanjikan untuk menstabilkan karbon atmosfer dalam bentuk padat. Di sini, hydrochar berstruktur nano yang didoping nitrogen, silikon, dan besi disintesis melalui karbonisasi hidrotermal biomassa satu langkah. Silika ditemukan membantu pembentukan ikatan C C, menghasilkan pembentukan lapisan karbon kristal. Setelah perawatan pirolisis, material yang dihasilkan memiliki nanopartikel besi valensi nol (Fe NP) yang tersebar merata yang dienkapsulasi dalam cangkang grafit atau oksida besi. Mekanisme pertumbuhan diusulkan, dan struktur serta mikrostruktur material dikarakterisasi menggunakan teknik multiskala yang saling melengkapi. Aktivitas elektrokatalitik terhadap reaksi reduksi oksigen dievaluasi dalam pengaturan tiga elektroda dan sel bahan bakar membran pertukaran proton, mencapai potensi onset dan kepadatan daya setinggi 0,76 V versus RHE dan 14,2 mW cm −2 , masing-masing. Stabilitas dinilai melalui uji tegangan yang dipercepat dan pengukuran beban berkelanjutan. Analisis pascastabilitas mengungkapkan bahwa aktivitas elektrokatalitik hanya dipertahankan oleh NP Fe yang dienkapsulasi grafit. Terakhir, sifat kapasitif bahan komposit diperiksa, dengan sampel terbaik menunjukkan kapasitansi dan energi spesifik masing-masing sebesar 60 F g −1 dan 8,1 Wh kg −1 .
1 Pendahuluan
Meningkatnya emisi CO2 global mengharuskan adanya evaluasi ulang terhadap pendekatan kita terhadap produksi material. Pada tahun 2022, peta jalan material berkelanjutan diterbitkan, [ 1 ] yang mempertimbangkan alternatif terbaik untuk produksi, konversi, dan penyimpanan energi. Material berbasis karbon sering kali menonjol, bersama dengan beberapa logam transisi, misalnya, besi. Saat ini, teknologi konversi energi bergantung pada penggunaan sumber daya mineral secara intensif, yang ekstraksi dan pemurniannya dikaitkan dengan emisi gas rumah kaca yang besar, polusi lahan, atau ketegangan geopolitik.
Dalam beberapa tahun terakhir, biochar, bahan seperti batu bara yang berasal dari biomassa, telah menarik perhatian global karena kemampuan penyimpanan karbon jangka panjangnya. [ 2 ] Biomassa tentu saja menampilkan efisiensi tertinggi dalam mengikat CO 2 dari atmosfer, meskipun untuk waktu yang terbatas karena dekomposisinya membebaskan jumlah atom karbon yang setara. Konversi biomassa menjadi biochar mengikat atom karbon dalam bentuk yang stabil dan, oleh karena itu, secara aktif berkontribusi pada penyerapan karbon. [ 3 ] Selain itu, biomassa dapat dipanen secara berkelanjutan, mudah tersedia dan dapat diakses di seluruh dunia, dan mudah dimasukkan ke dalam ekonomi sirkular. [ 4 ] Dalam beberapa dekade terakhir, karbonisasi hidrotermal (HTC) telah diperkenalkan kembali sebagai proses hemat energi untuk mengubah umpan biomassa menjadi hidrochar. Jika hidrochar ini telah digunakan secara luas untuk pengolahan udara atau air, [ 5 ] aplikasinya di bidang energi sangat terbatas karena konduktivitas elektroniknya yang buruk dan kurangnya aktivitas katalitik intrinsik untuk reaksi yang diinginkan. Perlakuan pasca, misalnya, pirolisis, biasanya digunakan untuk meningkatkan sifat material dalam kaitannya dengan aplikasi yang ditargetkan. Meskipun demikian, HTC menghadirkan kualitas yang unik berkat keserbagunaannya, yang memungkinkan seseorang untuk mencapai struktur yang unik dan doping heteroatom yang tinggi, dan terkenal karena meningkatkan hasil dan retensi heteroatom dalam struktur material selama perlakuan pasca berikutnya. [ 6 ]
Bahan karbon sangat serbaguna dan sifat fisikokimianya dapat disesuaikan secara ekstensif. Karbon dapat membentuk banyak struktur dan mikrostruktur dan cenderung membentuk ikatan kovalen yang kuat dengan berbagai heteroatom, misalnya, B, N, O, Si, P, atau S. Bahan karbon terdoping nitrogen telah dipelajari secara ekstensif untuk reaksi reduksi oksigen (ORR), yang sifat-sifatnya yang menjanjikan muncul dari modifikasi distribusi muatan pada atom C yang berdekatan dengan atom N, menghasilkan tempat adsorpsi untuk O 2 dan memfasilitasi disosiasinya. [ 7 ] Bahan karbon terdoping N juga terbukti memberikan jangkar yang baik untuk dukungan nanopartikel logam. [ 8 ] Sebaliknya, doping Si telah melihat sedikit penyelidikan, sebagian karena kesulitannya untuk dicapai sebagai akibat dari perbedaan besar dalam jari-jari atom antara atom C (77 pm) dan Si (117 pm). Meskipun demikian, Si berlimpah di Bumi, berada di biomassa (misalnya, sekam padi, bambu, tebu, atau sage) [ 9 ] atau di dalam tanah, yang membuatnya sangat menarik untuk desain bahan fungsional yang berkelanjutan. Selain doping, beberapa molekul yang mengandung Si telah dilaporkan mengkatalisis pembentukan ikatan karbon-karbon, yang sangat menarik untuk konversi biomassa pada suhu rendah. [ 10 – 12 ] Sejauh ini, doping silikon terutama berputar di sekitar modifikasi sifat tribologi film karbon amorf seperti berlian, [ 13 , 14 ] atau penyesuaian sifat titik kuantum karbon. [ 15 ]
Demikian pula, besi adalah unsur yang melimpah di Bumi, yang produksinya berbiaya rendah dan relatif ramah lingkungan dibandingkan dengan logam lain membuatnya sangat menarik. Elektrokatalis berbasis besi dapat dipisahkan menjadi dua kategori utama: katalis atom tunggal besi (SAC) dan nanopartikel besi (NP). Fe SAC biasanya terdiri dari kation Fe yang distabilkan dalam struktur inang (umumnya berbasis karbon) melalui koordinasinya dengan fungsi nitrogen piridin, membentuk spesies Fe-N4 yang terkenal. [ 16 ] Dalam konfigurasi ini, atom besi menunjukkan aktivitas elektrokatalitik tertinggi untuk ORR, tetapi juga jumlah situs aktif per satuan volume yang relatif rendah dan stabilitas yang buruk. [ 16 , 17 ] Di sisi lain, Fe NP dapat disiapkan dari berbagai metode sintesis. [ 18 ] Jika NP Fe awalnya dianggap tidak aktif untuk ORR, penelitian terbaru menemukan bahwa NP Fe, dan lebih khusus lagi NP besi bervalensi nol, yang terbungkus dalam cangkang karbon memiliki aktivitas elektrokimia yang menjanjikan, stabilitas tinggi, dan ketahanan tinggi terhadap keracunan permukaan. [ 19 ]
Di sini, kami menggunakan silika amorf bersama dengan fruktosa (atau daun maple) dan garam besi untuk menyiapkan lembaran karbon kodoping Si/N yang terorganisasi secara radial dengan besi yang terdispersi secara atomik, dari HTC satu langkah. Kami menunjukkan kesesuaian hidrokarbon yang disintesis untuk menghasilkan atmosfer reduktif selama pirolisis yang mengarah pada pembentukan NP besi bervalensi nol inti-kulit. Sifat elektrokimia bahan dinilai untuk ORR dalam media asam, baik dalam pengaturan tiga elektroda maupun sel bahan bakar, serta untuk aplikasi superkapasitor.
2 Hasil dan Pembahasan
2.1 Karakterisasi Fisikokimia
Bahasa Indonesia: Setelah karbonisasi hidrotermal, hidrokar FNS (Fruktosa–besi Nitrat–Silika) menunjukkan partikel bulat, berdiameter 0,2–1 μm, yang tersusun dari agregasi lembaran material tipis yang tampaknya berasal dari pusat partikel, yang menunjukkan kemungkinan mekanisme pertumbuhan radialnya ( Gambar 1a ). Beberapa nanostruktur berlapis dapat dengan mudah diamati dalam mikroskop elektron transmisi (TEM) (Gambar S1, Informasi Pendukung), meskipun mereka cepat terdegradasi di bawah berkas elektron terfokus. Difraksi elektron area terpilih mengonfirmasi sifat polikristalin dari struktur ini yang mungkin terkait dengan oksida grafit turbostratik yang rusak, menghadirkan jarak-d yang lebih besar daripada grafit biasa (≈4,70 versus 3,35 Å) karena adanya fungsi oksigen atau interkalasi, misalnya, atom Si. [ 20 , 21 ] Selain itu, beberapa partikel hidrochar bulat khas hadir dalam sampel, mudah dikenali dari permukaannya yang tampak halus (lih. Gambar S2, Informasi Pendukung). Kedua jenis partikel ini selanjutnya disebut sebagai “lembaran karbon terorganisir radial” (ROCS) dan “hidrochar”, masing-masing. Analisis spektroskopi sinar-X dispersif energi (EDX) dari dua kelompok partikel mengungkapkan bahwa jika ROCS tersusun dari campuran C, O, N, Si, dan Fe (lih. Tabel 1 , Gambar 1a ), partikel hidrochar hampir bebas dari atom Si dan Fe (lih. Gambar S2, Informasi Pendukung). Pemetaan EDX menunjukkan bahwa atom N, Si, dan Fe terdistribusi secara merata dalam ROCS, pada skala atom (lih. Gambar 1a ), yang menunjukkan kodoping N/Si/Fe. Namun, tidak dapat disimpulkan dari TEM hanya jika atom-atom ini terintegrasi ke dalam kisi karbon atau hadir sebagai fungsi permukaan. Hasil ini menyoroti kemampuan HTC dalam membentuk mikrostruktur unik dari biomassa. Dapat dikatakan bahwa, dalam kasus yang disajikan di sini, kemungkinan mekanisme bottom-up, yaitu, dari molekul fruktosa ke bahan karbon skala mikro, bertanggung jawab atas struktur spesifik bahan yang dilaporkan. Namun, sintesis serupa menggunakan daun maple sebagai pengganti fruktosa menghasilkan hasil yang hampir identik (lih. Gambar S3, Informasi Pendukung). Proses kompleks yang terjadi selama HTC telah dipelajari dalam penyelidikan sebelumnya [ 22 ] dan menghasilkan fragmentasi biomassa menjadi molekul kecil yang seharusnya sangat menguntungkan bagi penggabungan heteroatom ke dalam jaringan karbon. Beberapa penelitian menyelidiki konversi biomassa menjadi biochar dengan adanya silika melalui pirolisis langsung, [ 23 ] tetapi tidak ada yang mencapai hasil tersebut. Ini menunjukkan bahwa, selama HTC, umpan biomassa menghasilkan fungsi yang dapat bereaksi secara kimia dengan SiO 2, atau molekul berpusat pada Si tertentu yang dapat membantu pembentukan jaringan C C. Dua sampel disiapkan tanpa silika (ditandai FN) atau tanpa besi nitrat (FS) untuk mengidentifikasi apakah salah satu dari komponen ini bertanggung jawab atas pertumbuhan ROCS. Menariknya, tidak satu pun dari kedua sampel ini menunjukkan ROCS tetapi justru menunjukkan pemisahan unsur-unsur menjadi partikel hidrokarbon, oksida besi, dan silika (lih. Gambar S4, Informasi Pendukung). Hal ini menunjukkan bahwa interaksi spesifik terjadi ketika ketiga unsur tersebut hadir bersama-sama dalam reaktor batch, meskipun ini bukan fokus dari penelitian ini. Di samping pembentukan ROCS, penambahan silika ke reaktor batch sangat meningkatkan luas permukaan material, dari 137,5 menjadi 273,1 m 2 g −1 , dan volume pori total, dari 0,188 menjadi 0,377 cm 3 g −1 , untuk FN-800 dan FNS-800, masing-masing (lih. Gambar S5, Informasi Pendukung).

| Contoh ID | Kucing% | Si pada % [C:Si] | Fe dalam % [C:Fe] | N pada % [C:N] | O pada % [C:O] |
|---|---|---|---|---|---|
| FNS | 23,4 ± 4,8 | 12,5 ± 2,8 [1,9:1] | 9,8 ± 2,0 [2,4:1] | 4,0 ± 0,8 [5,9:1] | 50,3 ± 5,7 [0,5:1] |
| FNS-600 | 42,0 ± 1,1 | 7,6 ± 0,9 [5,5:1] | 6,6 ± 1,0 [6,4:1] | 4,1 ± 0,1 [10.2:1] | 39,7 ± 2,5 [1.1:1] |
| FNS-700 | 45,0 ± 1,0 | 7,1 ± 0,3 [6,3:1] | 5,4 ± 0,3 [8,3:1] | 4,6 ± 0,2 [9,9:1] | 37,9 ± 0,6 [1.2:1] |
| FNS-800 a) | 41,8 ± 9,7 | 11,1 ± 2,4 [3,8:1] | 6,6 ± 2,5 [6,4:1] | 3,5 ± 0,5 [11,9:1] | 37,1 ± 9,4 [1.1:1] |
| FNS-900 a) | 26,4 ± 8,5 | 14,1 ± 2,4 [1,9:1] | 12,0 ± 9,2 [2.2:1] | 2,2 ± 0,4 [12.2:1] | 45,3 ± 14,4 [0,6:1] |
a) Margin kesalahan yang besar disebabkan oleh bervariasinya jumlah Fe NP di area yang diukur.
Setelah pirolisis pada 600 dan 700 °C (Gambar 1b,c ), sampel tampaknya tidak mengalami modifikasi mikrostruktur yang signifikan. Namun, setelah pirolisis pada 800 °C (Gambar 1d ), material mengalami perubahan dramatis dengan munculnya nanopartikel besi (NP) dengan distribusi ukuran bimodal (12 ± 2 dan 81 ± 26 nm, lih. Gambar S6, Informasi Pendukung), yang terdistribusi secara merata pada permukaan material. Difraktogram sinar-X menunjukkan puncak yang jelas yang sesuai dengan Fe metalik untuk FNS-800 ( Gambar 2 ), selain puncak kecil yang dikaitkan dengan karbida besi (Fe3C ) dan oksida besi (Fe3O4 ) . Puncak lebar yang terletak dalam kisaran 20–30° dikaitkan dengan struktur karbon nanokristalin (misalnya, domain grafit kecil) dan/atau silikon dioksida. Karena kurangnya kepastian, puncak ini tidak akan dibahas lebih lanjut. Penampakan NP Fe mungkin berasal dari oksidasi struktur karbon dan demetalasi fungsi Fe berikutnya. Ini sebelumnya diamati dalam kondisi elektrokimia pada potensial oksidatif di mana spesies FeN4 mengalami demetalasi untuk membentuk Fe2 + . [ 24 ] Kami mendalilkan di sini bahwa proses seperti itu dapat terjadi pada degradasi termal struktur karbon. Selain itu, Varnell et al. menyarankan bahwa spesies N bertindak sebagai situs nukleasi untuk membentuk NP Fe, [ 19 ] yang dapat menjelaskan distribusi partikel yang seragam pada ROCS. Setelah pirolisis pada 900 °C (Gambar 1e ), NP besi lebih besar (137 ± 44 nm, lih. Gambar S6, Informasi Pendukung), yang menunjukkan proses difusi atom Fe pada permukaan ROCS ke titik nukleasi dan pematangan NP Fe kecil berikutnya menjadi yang lebih besar. Hipotesis ini diuji dengan mengurangi durasi pirolisis pada suhu 800 °C, dari 120 menjadi 60, 30, dan 15 menit. Hasilnya, diameter rata-rata NP Fe menurun menjadi 40 ± 13, 9,0 ± 1,6, dan 7,4 ± 1,5 nm, yang mengonfirmasi pematangan NP Fe selama pirolisis (lih. Gambar S7, Informasi Pendukung). Keberadaan NP besi metalik dalam sampel meskipun terpapar udara dan air menunjukkan pembentukan lapisan pasivasi atau enkapsulasi di dalam cangkang karbon. Pengamatan cangkang karbon grafit (ketebalan ≈5–20 nm, jarak antar lapisan 0,33 ± 0,02 nm, lih. Gambar S8, Informasi Pendukung), serta lapisan kaya oksigen di sekitar partikel Fe 0 (ketebalan ≈3–8 nm, lih. Gambar S8, Informasi Pendukung), menunjukkan bahwa kedua mekanisme tersebut terjadi. TEM juga menunjukkan bahwa Fe 0 yang dienkapsulasi karbonNP rata-rata lebih kecil daripada NP sejenisnya, kemungkinan karena relatif tidak sensitif terhadap pematangan. Hal ini menjelaskan distribusi ukuran partikel bimodal yang diamati untuk FNS-800, dengan partikel terkecil (12 ± 2 nm) terbungkus dalam cangkang karbon, sedangkan partikel yang lebih besar (81 ± 26 nm) terbungkus dalam cangkang FeOx. Difraksi sinar-X (XRD) yang dilakukan pada FNS-800 tepat setelah sintesis dan setelah 1 bulan terpapar udara tidak menunjukkan perubahan signifikan, yang mengonfirmasi stabilitas inti logam Fe (lih. Gambar S9, Informasi Pendukung).
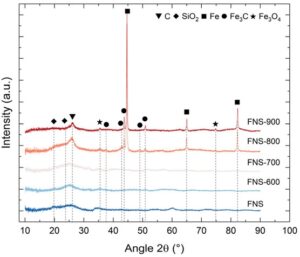
Kehadiran oksida besi hanya sebagai fase minoritas, seperti yang terlihat pada mikroskop elektronik dan XRD, disebabkan oleh atmosfer reduktif yang dihasilkan selama dekomposisi hidrokarbon, terutama melalui produksi CO dan H 2 in situ . [ 25 , 26 ] Meskipun sumber karbon hanya menghasilkan sejumlah kecil reduktor ini melalui dekomposisinya, kehadiran oksida logam (X), misalnya, oksida besi, menyebabkan reaksi sekunder yang mendukung evolusinya (Persamaan ( 1 ) hingga ( 4 )).

Dari sana, reduksi oksida besi menjadi besi metalik mengikuti jalur dari Persamaan ( 5 ) hingga ( 10 )

Analisis termogravimetri (TGA) digunakan untuk menyelidiki dekomposisi hydrochar di bawah N 2 , meniru kondisi pirolisis. Termogram yang diperoleh dengan FNS ditampilkan dalam Gambar 3a . Desorpsi air terjadi di bawah 100 °C, diikuti oleh dua wilayah yang menarik, 1) antara 350 dan 600 °C dan 2) antara 700 dan 900 °C. Wilayah pertama berhubungan dengan devolatilisasi hydrochar, [ 27 , 28 ] yaitu, transformasi bahan berbasis karbon menjadi gas, tar, dan residu padat. Pembentukan gas terkait dengan dekomposisi termal gugus fungsi dan pelepasan molekul ringan. Wilayah kedua berhubungan dengan evolusi H 2 , CO 2 , dan CO, menurut Persamaan ( 1 ) hingga ( 4 ). Penting untuk dicatat bahwa puncak-puncak di wilayah suhu tinggi ini terutama terjadi di hadapan FeO x dan hampir tidak ada untuk bahan karbon murni. [ 25 ] Mekanisme sintesis selaras dengan komposisi sampel yang diperoleh dengan EDX (Tabel 1 ). Rasio C:O kira-kira dua kali lipat setelah pirolisis pada 600 atau 700 °C dibandingkan dengan FNS yang tidak dipirolisis, yang menunjukkan penghapusan fraksi paling volatil dari hydrochar. Di atas 800 °C, fraksi C menurun karena evolusi CO dan CO 2 yang intens , disertai dengan peningkatan konsentrasi relatif Si dan Fe. Fraksi O meningkat secara signifikan pada 900 °C yang kemungkinan berasal dari oksidasi permukaan NP Fe di udara setelah sintesis.
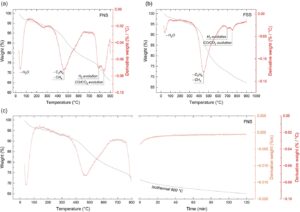
Rangkaian sampel kedua disiapkan dalam kondisi yang sama seperti rangkaian FNS x , hanya mengganti prekursor besi dari Fe(NO3 ) 3 · 9H2O menjadi FeSO4 · 7H2O , untuk menyelidiki efek ion lawan. Rangkaian ini selanjutnya dicatat sebagai FSS x (Fruktosa–besi Sulfat–Silika). Seperti dapat dilihat dari termogram yang dilaporkan pada Gambar 3b , FSS hanya menunjukkan puncak lemah pada wilayah 700–900 °C, sehingga menunjukkan relatif tidak adanya oksida besi untuk memulai reaksi yang dijelaskan pada Persamaan ( 1 ) dan ( 3 ). Analisis lebih lanjut dengan mikroskop elektron pemindaian (SEM) dan XRD (lih. Gambar S10, Informasi Pendukung) mengungkapkan bahwa seri FSS x sebagian besar gagal menghasilkan partikel besi setelah pirolisis, karena integrasi atom Fe yang buruk dalam bahan setelah HTC, dengan ≈2–4 wt% versus 20–25 wt% Fe dalam FSS dan FNS, masing-masing. Tidak ada nanostruktur spesifik yang diamati dalam TEM (seperti ROCS), dan atom C dan Si tampak terpisah antara partikel seperti hidrokarbon dan SiO 2. Hasil ini menggambarkan pentingnya atom N dan interaksinya dengan atom C, Si, dan Fe dalam pembentukan ROCS.
Spektrum Raman dipasang dengan dua puncak utama, pita G yang terkenal (≈1580 cm −1 ) dan pita D1 (≈1350 cm −1 ), masing-masing dikaitkan dengan vibrasi C sp 2 dalam bidang grafenik dan vibrasi mode pernapasan siklus aromatik dengan adanya cacat pada struktur grafenik. Penyesuaian Gaussian digunakan untuk memperhitungkan distribusi acak waktu hidup fonon dalam bahan karbon yang tidak teratur. [ 29 ] Tiga pita tambahan diperlukan untuk mencapai penyesuaian yang memuaskan (lih. Gambar 4a ), terletak di sekitar 1100 cm −1 (D4), 1500 cm −1 , dan 1750 cm −1 (D5). [ 30 ] Pita D4 biasanya muncul untuk bahan karbon yang kaya cacat. Kehadirannya dapat dikaitkan dengan ikatan sp 3 dalam karbon amorf tetrahedral. [ 31 ] Pita D5 dikaitkan dengan ikatan CO. [ 30 ] Menonjolnya bahu pada 1500 cm −1 (disebut Si C) tidak biasa untuk bahan karbon dan memang dikaitkan dengan peregangan ikatan Si C, [ 32 ] yang menunjukkan keberhasilan doping struktur karbon dengan atom Si. Rasio intensitas I Si–C / I G pada Gambar 4b tampaknya menunjukkan bahwa penggabungan atom Si dalam struktur karbon meningkat seiring dengan suhu pirolisis, sejalan dengan pengukuran EDX (lih. Tabel 1 ). Tidak ada puncak yang terkait dengan ikatan Si O yang diidentifikasi, biasanya terletak pada kisaran 800–1000 cm −1 , yang menunjukkan penggabungan total Si dalam struktur karbon dan/atau penghilangan silika selama langkah pencucian. Dengan cara yang sama, puncak triplet khas ≈800 cm −1 yang terkait dengan SiC kristal tidak diamati pada sampel apa pun. [ 33 ]
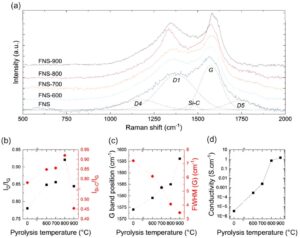
Rasio intensitas I D / I G (Gambar 4b ), serta posisi pita G (Gambar 4c ), meningkat seiring dengan suhu pirolisis. Pengamatan ini sesuai dengan transisi dari karbon amorf ke grafit nanokristalin. [ 29 ] Fraksi sp 3 dalam seri FNS x kemungkinan di bawah 15%, karena peningkatan lebih lanjut akan menghasilkan rebound posisi pita G ke nilai yang lebih tinggi, [ 29 ] yang diamati untuk sampel FSS yang tidak dipirolisis (lih. Gambar S10, Informasi Pendukung). Peningkatan kristalinitas seiring dengan peningkatan suhu pirolisis dikonfirmasi oleh pengukuran konduktivitas listrik (Gambar 4f ), mencapai nilai setinggi 1,44 S cm −1 untuk FNS-900. Sebagai perbandingan, konduktivitas karbon hitam komersial Vulcan XC-72 diukur pada 2,79 S cm −1 , sesuai dengan literatur. [ 34 , 35 ] Jumlah ikatan Si C, yaitu doping silikon, tampaknya meningkat seiring dengan suhu pirolisis hingga 800 °C, diikuti oleh penurunan tiba-tiba pada 900 °C (Gambar 4b ). Hal ini mungkin menunjukkan degradasi termal dari struktur material dan perubahan lingkungan atom Si, misalnya, gangguan ikatan Si C dan pembentukan ikatan Si Si.
2.2 Karakterisasi Elektrokimia
2.2.1 Reaksi Reduksi Oksigen Elektrokatalisis
Kurva polarisasi yang terekam pada sampel seri FNS x dalam 0,5 MH 2 SO 4 jenuh O 2 ( Gambar 5a ) menetapkan tren yang jelas dalam aktivitas dengan FNS-800 > FNS-900 > FNS-700 > FNS-600 > FNS. Hasil ini dapat dihubungkan dengan pembentukan partikel besi valensi nol yang dienkapsulasi, yang terbukti menjadi situs aktif untuk ORR. [ 19 ] Aktivitas FNS-900 yang lebih rendah kemungkinan disebabkan oleh diameter NP Fe yang lebih besar, yaitu, luas permukaan aktif yang lebih kecil. Voltammogram siklik (CV) mengungkapkan puncak reduksi dan oksidasi yang masing-masing terletak sekitar 0,5 dan 0,7 V RHE (Gambar 5b ). Dalam elektrolit jenuh N 2 (lih. Gambar S11, Informasi Pendukung), puncak redoks ini muncul sekitar 0,67 V RHE , yang dapat menunjukkan oksidasi dan reduksi berikutnya dari Fe 3 O 4 , yang diamati dengan XRD (Gambar 2 ), menjadi Fe 2 O 3 . [ 36 ] Potensi onset ORR, diukur sebagai potensi di mana kerapatan arus 0,3 mA cm −2 tercapai, serta kerapatan arus pertukaran, menunjukkan puncak untuk FNS-800, dengan nilai 0,76 V RHE dan 77 μA cm −2 , masing-masing (Gambar 5c ). Hasil ini sebanding dengan penelitian lain yang membahas elektrokatalis berbasis partikel besi dalam kondisi asam. [ 19 , 37 – 39 ] Menariknya, aktivitas serupa tercatat dengan sampel yang disiapkan dari daun maple MNS-800, dengan potensi onset ≈0,68 V RHE dan kerapatan arus pertukaran 34 μA cm −2 (lih. Gambar S2, Informasi Pendukung), meskipun dalam kasus ini, sampel optimal mungkin berada pada suhu pirolisis yang berbeda.

Mekanisme ORR diselidiki lebih lanjut pada FNS-800 dengan elektroda cakram cincin berputar (RRDE). Potensial cakram disapu dari 1 hingga 0 V RHE , sedangkan potensial cincin (Pt) dipertahankan pada 1,4 V RHE . Kurva polarisasi yang ditampilkan pada Gambar 5d menunjukkan deteksi H 2 O 2 dari ≈0,65 V RHE seterusnya, dan jumlah awal elektron n sekitar 2,7. Setelah adsorpsi O 2 pada situs katalitik (Persamaan ( 11 )), ORR dapat mengikuti dua jalur reaksi utama, yaitu proses 2 dan 4 elektron. Dalam proses 2 elektron, O 2 pertama-tama direduksi sebagian menjadi H 2 O 2 (Persamaan ( 12 )), sebelum direduksi lebih lanjut menjadi H 2 O (Persamaan ( 13 )). Dalam jalur 4 elektron, O 2 direduksi sepenuhnya menjadi H 2 O (Persamaan ( 14 )), tanpa membentuk zat antara H 2 O 2 . Rute ini biasanya lebih disukai karena zat antara H 2 O 2 dapat terlepas dari permukaan katalis sebelum membentuk H 2 O, yang dapat menyebabkan degradasi dini komponen sel melalui oksidasi kimia. Dalam kasus ini, nilai n menunjukkan campuran kedua jalur, dengan sedikit dominasi proses 2 elektron.
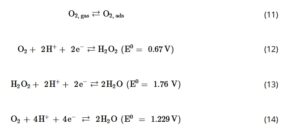
Prosedur uji stres yang dipercepat (AST) dilakukan pada suhu 25 °C untuk menilai stabilitas katalis dan kemungkinan perubahan dalam jalur reaksi. Selama 6000 siklus, yaitu 48 jam, jumlah elektron (diambil pada 0 V RHE ) secara bertahap meningkat hingga ≈3,0 (49% H 2 O 2 ) setelah sekitar 3000 siklus, sebelum secara bertahap turun kembali hingga ≈2,65 (68% H 2 O 2 ) setelah 6000 siklus (Gambar 5e ). Puncak tiba-tiba pada kurva disebabkan oleh gangguan sel dan tidak boleh diartikan sebagai perubahan aktivitas elektrokatalitik. Sepanjang AST, potensi onset ORR menunjukkan stabilitas luar biasa sekitar 0,72–0,74 V RHE , mirip dengan kerapatan arus pada 0 V RHE , yang stabil setelah ≈1000 siklus, dengan nilai dalam kisaran 0,85 mA cm −2 (Gambar 5f ). Menariknya, karakterisasi TEM pasca-AST mengungkapkan bahwa hanya NP Fe yang dienkapsulasi karbon yang tersisa dalam sampel (lih. Gambar S12, Informasi Pendukung). Ini menunjukkan bahwa 1) NP Fe yang dienkapsulasi FeO x tidak stabil dan terlarut dalam elektrolit asam dan 2) NP Fe yang dienkapsulasi karbon sendiri bertanggung jawab atas aktivitas elektrokatalitik menuju ORR. Selain itu, inti Fe 0 berada dalam kisaran 10 ± 2 nm, mirip dengan sebelum AST (12 ± 2 nm), sehingga menunjukkan stabilitas dan isolasi relatif inti dengan lingkungannya. Namun, cangkang karbon grafit kosong dapat diamati setelah AST, yang dapat disebabkan oleh oksidasi karbon (elektrokimia atau kimia karena adanya H 2 O 2 ) dan paparan inti Fe 0 ke elektrolit. Dalam jangka panjang, oksidasi inti Fe 0 dapat merugikan dalam konfigurasi sel bahan bakar, karena kehadiran H 2 O 2 dan Fe 2+ secara bersamaan diketahui mengoksidasi senyawa organik (reaksi Fenton). Konsentrasi Fe dalam sampel setelah AST diperkirakan dengan SEM-EDX (data tidak ditampilkan). Sesuai dengan pengamatan TEM, konsentrasi Fe sangat menurun, dengan hanya ≈0,1 at% (≈0,4 wt%) yang tersisa setelah AST, dibandingkan dengan ≈5,9 at% dalam material segar. Hasil ini mengungkapkan bahwa sebagian kecil dari NP Fe bertanggung jawab atas sebagian besar aktivitas katalitik, karena tidak ada penonaktifan signifikan yang terjadi selama AST.
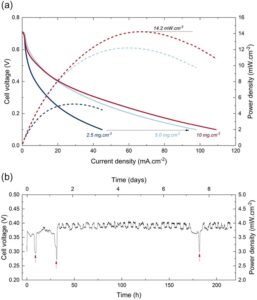
Akhirnya, sifat elektrokatalitik FNS-800 dipelajari dalam sel bahan bakar pada 50 °C untuk menilai kemungkinan penerapannya dalam perangkat konversi nyata. Tiga rakitan elektroda membran (MEA) disiapkan dengan pemuatan katalis yang berbeda pada katode, yaitu 2,5, 5,0, dan 10,0 mg cm −2 . Berbeda dengan logam mulia, pemuatan katalis saat ini tidak dibatasi oleh biaya atau ketersediaan bahan, melainkan oleh ketebalan elektroda yang, jika terlalu besar, menciptakan kerugian transportasi massa pada kepadatan arus yang tinggi. Dari Gambar 6a , jelas bahwa aktivitas total lapisan katalis mendapat manfaat dari pemuatan besar, dengan kepadatan arus pada 0,1 V meningkat dari 45 hingga 109 mA cm −2 untuk pemuatan 2,5 dan 10,0 mg cm −2 , masing-masing. Kepadatan daya mencapai maksimum 14,2 mW cm −2 , yang sekitar 15 kali lipat lebih rendah daripada kepadatan daya 20 wt% Pt yang ditopang pada material karbon yang diukur dalam pengaturan kami pada suhu 50 °C, dengan pemuatan 0,5 mg Pt cm −2 (lih. Gambar S13b, Informasi Pendukung). Seperti yang ditentukan dalam paragraf terakhir, hanya sebagian kecil dari NP Fe yang secara aktif mengkatalisis ORR dan akan sesuai dengan pemuatan tiga MEA yang diukur sebesar ≈10, 20, dan 40 μg cm −2 . Hasil ini sangat menjanjikan mengingat ruang untuk pengoptimalan masih tersedia dengan jenis material ini. Pengukuran stabilitas di bawah beban konstan (10 mA cm −2 ) dilakukan dengan MEA dengan pemuatan tertinggi. Bahan tersebut menunjukkan stabilitas yang luar biasa selama ≈215 jam pengukuran dengan tegangan sel bervariasi sekitar 0,38 V (Gambar 6b ). Beberapa fluktuasi tegangan terekam karena variasi suhu sel (≈52 ± 2 °C). Tiga periode pembanjiran terekam, yang mengakibatkan penurunan tegangan tiba-tiba (panah merah pada Gambar 6b ) karena lapisan katalis terhalang sebagian. Kurva polarisasi dan kerapatan daya (lih. Gambar S13a, Informasi Pendukung) yang terekam sebelum dan sesudah pengukuran stabilitas melaporkan penurunan ≈28% dalam kerapatan daya maksimum selama seluruh analisis. Investigasi mendalam melalui spektroskopi impedansi elektrokimia (EIS) (lih. Gambar S13c–e, Informasi Pendukung) mengungkap peningkatan lambat dalam resistansi transfer muatan katoda, dari 1,8 awal menjadi 2,0 Ω di akhir pengukuran, sementara transfer muatan anoda dan resistansi ohmik tetap stabil sekitar 0,08 dan 0,12 Ω. Secara keseluruhan, hasil ini sangat menjanjikan dan menunjukkan potensi tinggi NP Fe 0 yang dienkapsulasi karbon untuk mengkatalisis ORR dalam jangka waktu lama.
2.2.2 Kinerja Kapasitif
Kinerja kapasitif dari sampel yang disiapkan diselidiki dalam pengaturan tiga elektroda dengan N 2 -jenuh 0,5 MH 2 SO 4 , dengan melakukan voltametri siklik dan siklus pengisian–pelepasan muatan galvanostatik (GCD), dalam jendela potensial 0–1 V RHE . FNS-800 mengungguli semua sampel yang disiapkan lainnya ( Gambar 7a ), menyajikan perilaku kapasitif terbaik dengan CV hampir persegi panjang (Gambar 7b ) dan profil GCD kuasi-segitiga (Gambar 7c ). Material tersebut menunjukkan kapasitansi spesifik dan energi spesifik pada 1 mA cm −2 (≈0,86 A g −1 ) masing-masing sebesar 60 F g −1 dan 8,1 Wh kg −1 . Kemampuan laju FNS-800 diselidiki antara 1 dan 10 mA cm −2 (Gambar 7d ). Dalam rentang ini, kapasitansi dan energi spesifik bahan turun masing-masing sebesar ≈20% dan ≈44%. Kinerja FNS-800 yang baik dibandingkan dengan sampel lain dari seri FNS x (Gambar 7e ) menunjukkan peran penting yang dimainkan oleh perilaku pseudokapasitif dari NP Fe. Penurunan kapasitansi dan energi spesifik antara FNS-800 dan FNS-900 kemungkinan terkait dengan peningkatan diameter NP Fe, yaitu, penurunan luas permukaan (−40% di sini, data tidak ditampilkan).

Kinerja siklik FNS-800 dipelajari dengan siklus GCD dalam jendela potensial dari 0 hingga 1 V RHE , pada 1 A g −1 . Selama siklus, kapasitansi spesifik menunjukkan peluruhan yang hampir linear (Gambar 7f ) dan, setelah 2000 siklus, mempertahankan ≈83,6% dari kapasitansi awalnya, sementara energi spesifik menurun dari 5,9 menjadi 5,0 Wh kg −1 (−15%). Sementara itu, efisiensi Coulombic menurun dari ≈98,2% menjadi ≈86,8% (Gambar 7g ). Masuk akal untuk percaya bahwa, berbeda dengan sifat elektrokatalitik, baik FeO x – dan Fe NP yang dienkapsulasi karbon berperan dalam perilaku pseudokapasitif dari bahan yang disiapkan. Oleh karena itu, penurunan kapasitansi dapat dikaitkan dengan pelarutan bertahap dari FeNP yang dikapsulasi FeOx , seperti yang disebutkan di bagian sebelumnya.
3 Kesimpulan
Bahan karbon berstruktur nano yang dikodoping dengan N, Si, dan Fe berhasil disintesis melalui proses karbonisasi hidrotermal. Setelah pirolisis, heteroatom yang terdispersi secara atomik dalam struktur host membentuk NP Fe bervalensi nol, yang dienkapsulasi dalam cangkang grafit atau FeO x . Peningkatan suhu pirolisis dari 600 hingga 900 °C memperbaiki kristalinitas dan konduktivitas elektronik, meskipun suhu tertinggi juga menyebabkan pematangan NP yang tidak diinginkan. Analisis termogravimetri yang dilakukan dalam kondisi yang meniru perlakuan pirolisis terbukti menjadi alat yang menarik untuk memprediksi pembentukan nanopartikel besi dan menemukan kisaran suhu yang optimal. Kinerja elektrokatalitik untuk reaksi reduksi oksigen dievaluasi dalam sistem tiga elektroda dan sel bahan bakar membran pertukaran proton, mencapai potensi onset 0,76 V RHE dan kerapatan daya hingga 14,2 mW cm −2 . Pengukuran RRDE memberikan wawasan tentang mekanisme ORR, yang terjadi sebagai campuran jalur 2 dan 4 elektron. Uji stabilitas, termasuk siklus tegangan yang dipercepat (6000 siklus) dan pengukuran beban berkelanjutan (215 jam), menunjukkan ketahanan material yang menjanjikan. Analisis pasca stabilitas mengungkapkan bahwa aktivitas elektrokatalitik terutama disebabkan oleh NP Fe yang dienkapsulasi grafit, karena NP yang dienkapsulasi FeO x terlarut seiring waktu. Terakhir, sifat kapasitif komposit dievaluasi, dengan sampel dengan kinerja terbaik menunjukkan kapasitansi spesifik sebesar 60 F g −1 dan energi spesifik sebesar 8,1 Wh kg −1 .
4 Bagian Eksperimen
Persiapan Material Karbon-Silikon-Besi
Dalam sintesis tipikal, 1 g fruktosa (5,55 mmol, Sigma–Aldrich, ≥99%) dicampur dalam pelapis polipropiolakton (PPL) 50 mL (Shilpa Enterprises) dengan 0,3 eq garam besi (1,67 mmol), baik Fe(NO3 ) 3 · 9H2O ( Sigma–Aldrich, ≥98%) atau FeSO4 · 7H2O ( Honeywell, ≥99%), 0,3 eq silikon dioksida (1,67 mmol, Aldrich, 5–15 nm, 99,5%), sejumlah katalitik asam sitrat (5,5 mg, Sigma–Aldrich, ≥99%), dan 10 mL air deionisasi (Milli-Q, 18,2 MΩ cm). Campuran diaduk secara ekstensif dengan tangan sebelum menutup bejana dan menaruhnya dalam autoklaf hidrotermal baja tahan karat. Autoklaf ditutup pada ragum untuk mencegahnya terbuka selama reaksi karena ekspansi/kontraksi termal. Autoklaf ditempatkan dalam oven Memmert 100–800 dan dipanaskan hingga 280 °C (1,5 °C min −1 ) selama 24 jam. Campuran yang dihasilkan disaring vakum pada filter membran polipropilena (0,2 μm, Merck), dan residu padat (hidrokarbon) dicuci terlebih dahulu dengan etanol terdenaturasi (etanol 91,2%, aseton 3%, Anora Industrial) dan kemudian dengan air deionisasi. Hidrokarbon dikeringkan dalam oven vakum (Heraeus VTR 5022) selama sekitar 1 jam. Terakhir, hidrokarbon tersebut dipirolisis di bawah N 2 (250 mL min −1 ) selama 2 jam pada suhu 600–900 °C (4–5 °C min −1 ) dalam tungku tabung Nabertherm RS80/500/11.
Karakterisasi Fisikokimia
Resistivitas listrik diukur dengan sel khusus dalam konfigurasi dua probe. Sejumlah kecil material dimasukkan di antara dua piston baja tahan karat (diameter 10 mm), dibungkus dalam jaket polimer nonkonduktif, dan ditekan hingga mencapai ≈1,3 t cm −2 . Serangkaian arus listrik disuntikkan di antara dua piston menggunakan unit pengukuran sumber Keithley (Keithley 2400) dan tegangan yang dihasilkan dicatat. Resistivitas listrik material R (Ω) diubah menjadi resistivitas spesifik ρ (Ω cm) menggunakan persamaan berikut
![]()
di mana A adalah luas penampang sampel (cm 2 ) dan l adalah ketebalan sampel (cm). Resistansi material dikoreksi dari resistansi sel kosong, biasanya sekitar 0,3 Ω. Ketebalan lapisan material, dalam kisaran 2–5 mm, diukur menggunakan pengukur ketebalan, dengan membandingkan dimensi sel dengan dan tanpa sampel di bawah tekanan yang sama. Spektroskopi Raman dilakukan pada Renishaw inVia dengan panjang gelombang laser 532 nm, dalam kisaran 200–2000 cm −1 . Sebelum pengukuran, sampel diposisikan pada slide mikroskop kaca dan dikompresi dengan lembut untuk mencapai permukaan datar. Garis dasar dikurangi dari semua spektrum sebelum analisis. Spektrum didekonvolusi dengan kurva Gaussian dan Lorentzian, sesuai dengan literatur, [ 30 ] dan pemasangan dioptimalkan dengan meminimalkan perbedaan absolut antara data eksperimen dan data terhitung. TEM dilakukan dengan JEOL JEM-2800 yang dioperasikan pada 200 kV. Sampel didispersikan dalam etanol (Etax Aa, Anora, >99,5 wt%) sebelum dituang pada kisi karbon berlubang (AGS147-3, Agar Scientific). Analisis EDX diperoleh dengan detektor kecepatan tinggi pada peralatan yang sama. XRD dilakukan pada X’pert Pro MPD ( λ = 1,5406 Å). Komposisi sampel dianalisis terhadap basis data Pusat Internasional untuk Data Difraksi (ICDD). TGA dilakukan pada TGA 5500 dari TA Instrument dalam atmosfer inert (N 2 , 25 mL min −1 ). Sekitar 10 mg material dimasukkan ke dalam wadah alumina, yang ditahan di tungku oleh panci Pt. Termogram direkam dengan peningkatan pemanasan 3 °C min −1 .
Karakterisasi Elektrokimia
Elektrokatalis dipelajari dalam pengaturan tiga elektroda. Tinta yang mengandung katalis disiapkan dengan mencampur 5 mg katalis dengan 100 μL air deionisasi, 100 μL etanol (Etax Aa, Anora, >99,5 wt%), dan 20 μL larutan resin perfluorinasi yang mengandung Nafion 1100 W (5 wt%, Sigma–Aldrich). Setelah pengadukan menyeluruh, 10 μL tinta diteteskan pada ujung karbon kaca (GC) 0,196 cm2 dan dibiarkan kering dalam kondisi sekitar. Setelah kering, GC dimasukkan ke dalam dudukan Teflon dan direndam dalam 0,5 MH2SO4 panas selama sekitar 30 detik untuk mengurangi sifat hidrofobik lapisan katalis . Pengaturan elektrokimia terdiri dari tiga elektroda yang dimasukkan ke dalam sel kaca. Elektroda kerja dipasang pada rotator (kontrol kecepatan Pine MSRX), berputar pada kecepatan 1600 rpm, elektroda lawan adalah kawat platinum, dan elektroda referensi adalah elektroda hidrogen reversibel (RHE) (Gaskatel, Hydroflex). Analisis dilakukan dalam elektrolit 0,5 MH 2 SO 4 (Merck, Titripur) pada 25 °C. Nitrogen digelembungkan selama 30 menit sebelum pengukuran pertama untuk memastikan penghilangan O 2 yang terlarut dalam elektrolit. Pengukuran diujicobakan dengan potensiostat Autolab (PGSTAT128 N). Semua sampel dikarakterisasi mengikuti prosedur yang sama. Antara 30 dan 50 CV (25 mV s −1 ) direkam di wilayah 0,0–0,6 V RHE hingga 10 siklus berturut-turut tidak menunjukkan perubahan signifikan. EIS potensiostatik dilakukan pada potensial sirkuit terbuka (OCP) untuk memperkirakan kontribusi iR , dalam rentang frekuensi 10 kHz hingga 100 mHz dengan fluktuasi AC sebesar 10 mV. Dua CV direkam dalam rentang yang diperluas dari 0,0 hingga 1,2 V RHE , pada laju pemindaian 50 mV s −1 . Setelah langkah ini, elektrolit dijenuhkan dengan O2 ( Woikoski, ≥99,999%) selama 30 menit, dan aliran gas dipertahankan selama karakterisasi berikutnya. EIS pada OCP (≈0,8–0,9 V RHE ) dan CV antara 0,0 dan 1,2 V RHE diulang dengan cara yang sama seperti pada elektrolit jenuh N2 . Voltammogram sapuan linear (LSV) direkam dari 1,0 hingga 0,0 V RHE pada 10 mV s −1 setelah 10 detik stabilisasi pada 1,0 V RHE . Potensi onset ditentukan dari kurva LSV, diambil sebagai potensi di mana kerapatan arus melebihi −0,3 mA cm −2 . Kerapatan arus pertukaran i 0 dihitung dari pemasangan kurva LSV dengan Persamaan Butler-Volmer ( 16 ) dalam rentang kelebihan potensial (η) dari 0 hingga −100 mV.
![]()
di mana i adalah kerapatan arus, α adalah koefisien transfer muatan, F adalah konstanta Faraday, T adalah suhu absolut, dan R adalah konstanta gas universal. Penentuan i 0 dilakukan dengan meminimalkan kesalahan antara data eksperimen dan data yang dihitung. Setelah karakterisasi kinetika ORR, elektrolit dijenuhkan sekali lagi dengan N 2 selama 30 menit untuk mengukur perilaku kapasitif bahan. CV direkam antara 1,0 dan 0,0 V RHE pada 10, 25, 50, dan 100 mV s −1 . Arus I (A) diubah menjadi kapasitansi C (F) melalui Persamaan ( 17 )
![]()
di mana ν adalah laju pemindaian (V s −1 ). Kapasitansi spesifik C sp dihitung dengan membagi kapasitansi rata-rata selama CV penuh (pada 100 mV s −1 ) dengan massa material m yang diendapkan pada GC (≈0,23 mg). Kurva pengisian–pelepasan galvanostatik (GCD) direkam antara 0,0 dan 1,0 V RHE pada rapat arus 1, 2, 5, dan 10 mA cm −2 , yaitu, ≈0,86, 1,72, 4,3, dan 8,6 A g −1 . Kapasitansi spesifik C sp dan energi spesifik E dihitung menggunakan Persamaan ( 18 ) dan ( 19 ), masing-masing
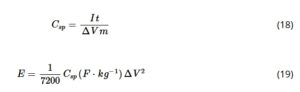
di mana I adalah arus pelepasan konstan, t adalah waktu pelepasan, m adalah massa material, dan Δ V adalah jendela potensial. Stabilitas dinilai pada material baru dalam elektrolit jenuh N 2 dengan melakukan 2000 siklus GCD pada 1 A g −1 . Efisiensi Coulombik CE dari setiap siklus dihitung sebagai berikut:
![]()
di mana q charge/discharge adalah muatan listrik yang disimpan/dilepas (C). Pengukuran elektroda cakram cincin berputar (RRDE) dilakukan dengan RRDE Cakram Tetap E6R2 (Pine Research, luas cakram: 0,2376 cm 2 , luas cincin: 0,2356 cm 2 ), yang menunjukkan efisiensi pengumpulan sebesar 38,3%. Material pertama-tama dimasukkan ke dalam prosedur pengkondisian dalam elektrolit jenuh N 2 yang terdiri dari 30 CV antara 0,6 dan 0,0 V RHE pada 25 mV s −1 , diikuti oleh EIS pada OCP. Setelah menjenuhkan elektrolit dengan O 2 selama 30 menit, prosedur uji tegangan dipercepat dimulai, yang terdiri dari pergantian 25 siklus CV dari 1,0 hingga 0,2 V RHE (100 mV s −1 ) dengan satu LSV dari 1,0 hingga 0,0 V RHE (5 mV s −1 ). Potensial cincin dijaga pada 1,4 V RHE , cukup tinggi untuk mengoksidasi H 2 O 2 (E 0 = 1,14 V RHE ). Fraksi H 2 O 2 yang dihasilkan oleh elektrokatalis dan jumlah elektron n yang sesuai dihitung menurut Persamaan ( 21 ) dan ( 22 ), masing-masing.

di mana J R adalah arus ring (A), J D adalah arus disk (A), dan N adalah efisiensi pengumpulan. Pengukuran sel bahan bakar dilakukan dalam sel komersial (Fuel Cell Technology, Inc.) pada suhu 50 °C dan kelembaban relatif (RH) gas sebesar 100%. H 2 (50 mL min −1 , Woikoski, ≥99,999%) dan O 2 (50 mL min −1 , Woikoski, ≥99,999%) diumpankan pada sisi anoda dan katoda, masing-masing. Pengukuran dilakukan dengan potensiostat Autolab (PGSTAT100). Tinta katalis katoda disiapkan dengan mencampur FNS-800 dengan air deionisasi, iso-propanol (Honeywell, Chromasolv, ≥99,9%), dan larutan Nafion (Sigma–Aldrich, 5 wt%). Kuantitas dipilih dengan asumsi efisiensi penyemprotan 45%, sehingga pemuatan katalis mencapai 2,5, 5,0, atau 10,0 mg cm −2 , dan konsentrasi Nafion dalam lapisan katalis konstan pada 30 wt%. Kuantitas air deionisasi dan isopropanol ditentukan dari data empiris, dengan rasio volume terhadap massa (μL mg −1 katalis) masing-masing 3 dan 20. Tinta katalis anoda disiapkan secara identik, menggunakan 20 wt% Pt@C (Premetek) dan bertujuan pada pemuatan 0,5 mg Pt cm −2 . MEA disiapkan dengan menyemprotkan tinta katalis pada kedua sisi membran Nafion 20 cm 2 115 (Ion Power), menggunakan pola persegi 5,29 cm 2 . Membran dikeringkan selama 2 jam pada suhu 80 °C dalam oven vakum (Heraeus) dan ditimbang sebelum dan setelah setiap penyemprotan untuk mengukur muatan katalis aktual pada setiap sisi membran. MEA ditekan di bawah tekanan 5 t pada suhu 130 °C selama 2 menit dan disimpan dalam air deionisasi sebelum dimasukkan ke dalam sel, diapit di antara dua lapisan difusi gas (Sigracet 22 BB).